Dr. Weeks’ Comment: These studies suggest that needing to use insulin DOUBLES your risk for pancreatic cancer another great reason to eat rich, exercise, sleep deeply and avoid diabetes.
Insulin promotes pancreatic cancer: evidence for endocrine influence on exocrine pancreatic tumors.
Source
Department of Surgery, Ohio State University, Columbus 43210, USA.
Abstract
Type-II diabetes is a risk factor for pancreatic cancer. In addition, diabetic patients present with more advanced tumors and have shortened survival compared to stage-matched counterparts. We hypothesize that the diabetic endocrine milieu, particularly elevated plasma insulin, favors pancreatic cancer growth. This study examines six human pancreatic cancer cell lines for the presence of insulin receptors and the influence of insulin on tumor proliferation. Classical competitive binding assays are performed using [125I insulin. Cell proliferation assays are conducted over 3 days on cultured cell lines (n = 6 replicates) with increasing concentrations of insulin. Insulin receptors are demonstrated on all six cell lines and dose dependent increases in cell proliferation (15-120% of control) are demonstrated in response to insulin. Patients with type-II diabetes hypersecrete insulin. The presence of high-affinity insulin receptors and dose dependent increases in pancreatic cancer cell proliferation with insulin supports the hypothesis that insulin may be an important tumor growth promoter in diabetes, particularly if paracrine mechanisms are involved. Additional study is required to determine whether other islet peptides altered in diabetes influence tumor growth and whether elevated plasma insulin favors pancreatic cancer induction.
AND
Pancreatic Cancer Tied to Insulin Woes
WebMD Health News
Dec. 13, 2005 — A new study links insulin problems to greater odds of developing pancreatic cancer.
Insulin is a hormone. It’s made by the pancreas and it’s necessary for the body to be able to use blood sugar (glucose) for fuel.
Insulin resistance occurs when there are elevated levels of glucose despite the presence of insulin. The body becomes resistant to the insulin available and strives to make more in order to counter the elevated glucose levels. Insulin resistance can lead to type 2 diabetes.
The news about insulin and pancreatic cancer appears in The Journal of the American Medical Association. The National Cancer Institute’s Rachael Stolzenberg-Solomon, PhD, and colleagues worked on the report.
Pancreatic cancer is the No. 4 cause of U.S. cancer deaths, states the American Cancer Society’s web site.
Male Smokers Studied
Data came from a study of male smokers in Finland that looked for the future incidence of cancers.
More than 29,000 men took part in the study, which started in 1985.
Stolzenberg-Solomon’s team focused on 169 men who developed pancreatic cancer. For comparison, they also included 400 randomly chosen men from the study who didn’t develop pancreatic cancer. The men were followed until December 2001.
Key Information
The study hinged on blood samples given by the men at least five years before any of them were diagnosed with pancreatic cancer.
Some of the men’s blood samples showed higher levels of blood glucose, insulin resistance, and high insulin levels. Those men were more likely to later develop pancreatic cancer than those without insulin problems.
The odds of getting pancreatic cancer were worse for men with type 2 diabetes and the highest insulin levels. They were twice as likely to develop pancreatic cancer as those with the lowest insulin levels, the study shows.
The men’s age, smoking status, and BMI (body mass index) didn’t change the results.
Study’s Limits
The study doesn’t prove that insulin problems cause pancreatic cancer.
The researchers note that scientists disagree about which comes first: undetected pancreatic cancer or insulin problems.
The study only included male smokers. But the findings are consistent with other research that included women and nonsmokers, write Stolzenberg-Solomon and colleagues.
They add that while their results support the idea that insulin problems are connected to pancreatic cancer, confirmation is needed from other studies.
Prevention Possibility
Avoiding insulin problems “could possibly impact pancreatic cancer development, as well as other cancer and chronic disease,” write the researchers. But they didn’t test that theory.
How do you avoid insulin problems? Stolzenberg-Solomon and colleagues mention three lifestyle changes:
- Lose extra weight
- Increase physical activity
- Improve your diet (such as eating less saturated fat, which is found in animal-based foods and some oils)
he Intracellular Mechanism of Insulin Resistance in Pancreatic Cancer Patients1
–Author Affiliations
Department of Biomedical Sciences, Creighton University School of Medicine (J.Li., J.A.K., T.E.A.), Omaha, Nebraska 68178; and Department of Surgery, Karolinska Institute at the Huddinge Hospital (L.S., J.P., J.La.), Stockholm S14186, Sweden
- Address correspondence and requests for reprints to: Thomas E. Adrian, Ph.D., F.R.C.Path., Department of Biomedical Sciences, Creighton University School of Medicine, Omaha, Nebraska 68178. E-mail: tadrian@creighton.edu.
Abstract
The diabetes that frequently occurs in pancreatic cancer patients is characterized by profound peripheral insulin resistance. The intracellular mechanism of this insulin resistance was investigated in skeletal muscle biopsies from pancreatic cancer patients with or without diabetes and control subjects.
Insulin receptor (IR) binding, tyrosine kinase activity, IR messenger RNA (mRNA), IR substrate-1 content, GLUT-4, and GLUT-4 mRNA content were all normal in pancreatic cancer patients. In contrast, multiple defects in glycogen synthesis were found in pancreatic cancer patients, especially in those with diabetes. Glycogen synthase I activity, total activity, and mRNA levels were significantly decreased in pancreatic cancer patients compared with controls. The fractional velocity of glycogen synthase was decreased only in the diabetic pancreatic cancer group. Glycogen phosphorylase a and b activities were increased in diabetic pancreatic cancer patients, but glycogen phosphorylase mRNA levels were not significantly different. The insulin resistance associated with pancreatic cancer is associated with a post-IR defect, which impairs skeletal muscle glycogen synthesis and glycogen storage.
UP TO 80% of pancreatic cancer patients have diabetes or impaired glucose tolerance at the time of cancer diagnosis (1, 2). Recent studies suggest that diabetes is most likely caused by the presence of the pancreatic tumor, rather than a risk factor for the cancer (3, 4, 5). The impaired glucose metabolism associated with pancreatic cancer is characterized by relatively normal insulin secretion and markedly reduced insulin sensitivity, indicating the presence of peripheral insulin resistance (6, 7, 8, 9). An improvement in both whole body glucose metabolic capacity and peripheral insulin sensitivity after pancreatectomy in pancreatic cancer patients has been reported, suggesting that the insulin resistance is caused by the presence of the pancreatic tumor (6, 10).
Skeletal muscle is the most important target tissue for insulin in glucose disposal (11). Peripheral insulin resistance may result from any defect that alters the action of insulin at the cellular level of target tissues (12, 13). The binding of circulating insulin to its specific receptor is the first step for insulin action (14). The receptor tyrosine kinase and insulin receptor (IR) substrates are crucial for insulin signal transduction from IR on the membrane into the postreceptor pathway in the cell (15). The final step of insulin action in skeletal muscle is to stimulate glucose transport and glycogen synthesis. Glucose uptake is mediated by facilitated diffusion through plasma membrane glucose transporters (16). The GLUT-4 transporter is insulin sensitive and responsible for insulin-stimulated glucose transport (16). Nonoxidative glucose disposal (mainly glycogen synthesis in skeletal muscle) has been considered as the primary site of insulin resistance (17). Glycogen synthase is the rate-limiting enzyme in the glycogen synthetic pathway (18). It has been reported that skeletal muscle glycogen synthase activity correlates with insulin-stimulated glucose disposal in vivo, indicating it plays a major role in nonoxidative glucose disposal (18). On the other hand, skeletal muscle glycogen content is also regulated by glycogen phosphorylase, which catalyzes glycogenolysis.
The intracellular mechanism of peripheral insulin resistance in diabetes associated with pancreatic cancer is unknown. To investigate the mechanism involved, the present studies were designed to measure IR binding, receptor tyrosine kinase activity, IR substrate-1 (IRS-1) content, glycogen synthase activity, glycogen phosphorylase activity, and insulin response glucose transporter (GLUT-4) content in skeletal muscles from pancreatic cancer patients with or without diabetes and in healthy controls.
Material and Methods
Human studies
The groups studied included eight pancreatic cancer patients with diabetes, seven pancreatic cancer patients with normal glucose tolerance, and nine patients with nonmalignant esophageal disease (controls). The clinical characteristics of these groups are shown in Table 1⇓. The pancreatic cancer patients were stented before this procedure. The division of pancreatic cancer patients into normal glucose tolerance and a diabetic group was made according to World Health Organization criteria (19). Therapy with oral hyperglycemic agents was stopped at least 2 weeks before surgery, and all diabetic patients were treated with insulin during the period before surgery. Surgical skeletal muscle biopsies (∼300-500 mg) were taken from the rectus abdominis muscle, just after the induction of anesthesia for laparotomy, from patients with pancreatic cancer and from the control subjects undergoing elective surgery. These skeletal muscle biopsies were snap-frozen in liquid nitrogen immediately after the biopsy and stored at −80 C for subsequent extraction and assay. All studies were approved by the local ethics committee of Linköping University Hospital, and informed consent was obtained from each patient.
Clinical characteristics of pancreatic cancer patients and controls
In vitro biochemical assays
Solubilization and partial purification of skeletal muscle insulin receptors was carried out using modification of a previously described technique (20).
The WGA-agarose eluate (25 μL) was incubated with A14[ 125I] insulin (∼30,000 cpm, specific activity 2000 Ci/mmol; Amersham Pharmacia Biotech, Arlington Heights, IL) without or with various concentrations of unlabeled insulin (10−11-10−6 M) at 4 C for 18 h. Binding in the presence of 1 μM unlabeled insulin was considered to be nonspecific. The IR binding capacity and receptor binding affinity in each receptor preparation was determined by Scatchard analysis using the LIGAND program (National Institutes of Health, Bethesda, MD).
IR tyrosine kinase activity was measured by using synthetic poly Glu-Tyr (4:1) as the substrate, as described previously (20).
IRS-1 protein level was determined by Western blot analysis. Protein concentration was measured by the Bradford method (Bio-Rad Laboratories, Inc., Richmond, CA). Equal amounts of solubilized skeletal muscle protein (20 μg) measured by the Bradford method were resolved by SDS-PAGE and transferred to polyvinylidene difluoride membrane. Membranes were immunobloted with anti-IRS-1 antibodies and horseradish peroxidase-labeled antirabbit antibodies, and IRS-1 bands were detected with a horseradish peroxidase/chemiluminiscense system (ECL; Amersham Pharmacia Biotech). Protein bands were quantified by densitometric scanning and expressed as OD/μg protein.
Skeletal muscle glycogen synthase and glycogen phosphorylase activities in crude homogenate was measured by the methods of Bak et al. (20) and Gilboe et al. (21), respectively.
Total content of the insulin-responsive glucose transporter GLUT-4 in the skeletal muscle tissue was measured in a crude membrane preparation, as described by Lund et al. (22). Skeletal muscle membrane preparations (50 μg/lane) were subjected to 10% SDS-PAGE and electrophoretically transferred to a nitrocellulose membrane. The membrane was then immunoblotted with the monoclonal antibody 1F8 (5 μg/mL; Genzyme Co., Cambridge, MA) and125I-labeled sheep antimouse f(ab”²)2 fragment second antibody (∼ 20,000 cpm/100 μL, specific activity 19μ Ci/μg; Amersham Pharmacia Biotech). The specific GLUT-4 protein band was detected and quantified using a phosphorimager (Bio-Rad Laboratories, Inc. Hercules, CA).
Quantitative RT-PCR
Total skeletal muscle tissue RNA was extracted using the guanidine thiocyanate water-saturated phenol extraction method (23). The integrity of the RNA was evaluated by electrophoresis using a nondenaturing 1% agarose gel.
IR, glycogen synthase, glycogen phosphorylase, and GLUT-4 messenger RNA (mRNA) in skeletal muscle samples were quantified by competitive RT-PCR. DNA primers used in RT-PCR were synthesized using an automated DNA synthesizer (392 DNA synthesizer; PE Applied Biosystems, Foster City, CA). The primers for RT-PCR are shown in Table 2⇓.
Primers for RT-PCR
Heterologous competitor DNA fragments were generated using composite primers that contain upstream or downstream primers for the target sequence linked to the upstream or downstream primers annealing to a heterologous DNA fragment of the Luciferase gene. PCR was performed using Gene Amp PCR Reagent Kit (Perkin-Elmer Corp., Foster City, CA) at 95 C, 15 sec, and 59 C, 30 sec, for 35 cycles. Then, the PCR products were purified using a Geneclean II Kit (Bio 101 Inc., La Jolla, CA) and cloned using the Original TA Cloning Kit (Invitrogen Co., San Diego, CA). The cloned plasmid DNA was extracted using an alkaline lysis mini-prep procedure, and the insert was sequenced (24). In vitro transcription was performed by incubation of linearized plasmid DNA (5 μg) with 1× T7 transcription buffer, 10 mM DTT, 100 U RNase inhibitor, 0.5 mMATP, 0.5 mM GTP, 0.5 mM TTP, 0.5 mM UTP, and 50 U T7 polymerase (Gibco BRL, Life Technologies, Inc., Gaithersburg, MD) in a final volume of 100 μL for 2 h at 37 C. The transcribed complementary RNA was DNase treated and quantified by absorbance at 260 nm.
Endogenous insulin receptor, glycogen synthase, glycogen phosphorylase, and GLUT-4 mRNA in skeletal muscle were quanitifed by coamplifying the cRNA competitor at various concentrations with target mRNA in RT-PCR using GeneAmp RNA PCR Kit (Perkin-Elmer Corp., Foster City, CA). RT was carried out in a 20 μL reaction mixture with 1μ g total RNA and various concentrations of competitive cRNA. PCR was then performed in a 100 μl mixture at 95 C, 105 sec for one cycle, at 95 C, 15 sec, 59 C, 30 sec for 35 cycles, and at 72 C for 7 min. The resulting ethidium bromide-stained DNA bands were visualized under ultraviolet light, and the density of each band was quantified using the Molecular Analyst program (Version 1.4, Bio-Rad Laboratories, Inc., Hercules, CA). The log of the ratios of the target products/competitor products was plotted against the log of the amount of competitor cRNA in each reaction, to yield the equivalence point between target mRNA and cRNA. The amount of IR, GLUT-4, glycogen synthase, and phosphorylase mRNA in skeletal muscle tissue is presented as attomole of mRNA in 1 μg total RNA sample.
Statistical methods
The results are presented as mean ± SEM. Differences between groups were determined using ANOVA with Bonferroni correction for multiple comparisons. P values less than 0.05 were considered statistically significant.
Results
IR binding and IR mRNA
The competition-inhibition binding curves were similar among pancreatic cancer groups with and without diabetes and control group as shown in Fig. 1A⇓. There were no significant differences in IR density, receptor binding affinity, or IR mRNA levels between the controls and cancer groups (Table 3⇓, Fig. 1B⇓).

Characterization of IR binding and receptor mRNA in skeletal muscles of pancreatic cancer patients and controls. A, Insulin binding to partially purified IRs from skeletal muscle biopsies. The column eluate (25 μl) was incubated with A14[125I] insulin (∼30,000 cpm, specific activity 2000 Ci/mmol) in the absence or the presence of various concentrations of unlabeled insulin (10−11-106 M) in a total volume of 150 μl at 4 C for 18 h. B, Representative gel electrophoresis of competitive RT-PCR analysis of IR mRNA. Lanes 1-6 represent IR RT-PCR products from 1 μg total RNA in the presence of IR competitive cRNA mimic (Lane 1, 989 amol; Lane 2, 32 amol; Lane 3, 10 amol; Lane 4, 3.2 amol; Lane 5, 1.0 amol; Lane 6, 0.3 amol). The top 326-bp DNA band is a product of the IR competitive cRNA, and the bottom 252-bp DNA band is an amplification product of the endogenous IR mRNA. IR mRNA levels were quantified by competitive RT-PCR. The results are presented as the mean ± SEM.
Skeletal muscle insulin receptor densities (Bmax), affinities (Kd) and insulin receptor tyrosine kinase activities in pancreatic cancer patients and controls
IR tyrosine kinase activity and IRS-1 content
Insulin dose-dependently stimulated receptor tyrosine kinase activity with maximal stimulation at 300 nM (Fig. 2A⇓). There were no significant differences in maximal receptor tyrosine kinase activities or concentrations of insulin for half-maximal activation of tyrosine kinase activity between groups (Table 3⇑). IRS-1 protein contents were found to be similar between control subjects and pancreatic cancer patients with or without diabetes (Fig. 2B⇓).

A, Tyrosine kinase activity of lectin-purified IRs from skeletal muscles of pancreatic cancer patients with and without diabetes and control subjects. Aliquots of lectin column eluate (60 μl) were incubated without and with various concentrations of insulin (0.3-300 nM) for 30 min at room temperature. Phosphorylation was carried out in the presence of [γ-32P] ATP (30 μM, specific activity 1 μCi/nmol) and synthetic substrate poly Glu4-Tyrl for 20 min at 4 C. The amount of 32P incorporated was normalized to the binding capacity calculated from Scatchard plot of insulin binding data. B, Quantification of IRS-1 protein in skeletal muscles from pancreatic cancer patients and controls. A representative Western blot of solubilized muscle proteins. Skeletal muscle biopsies were homogenized, and equal amounts of proteins were resolved in SDS-PAGE, transferred to PVDF membranes, immunoblotted with anti-IRS-1 antibodies and detected with a horseradish peroxidase/chemilluminescense system. Protein bands were quantified by densitometric scanning and expressed as OD/μg protein ± SEM. The results are presented as the mean± SEM.
Skeletal muscle glycogen synthase activity and mRNA
Basal glycogen synthase activity measured in the absence of glucose 6-phosphate was significantly decreased in diabetic pancreatic cancer patients compared with controls (Fig. 3A⇓, Table 4⇓). Glycogen synthase I activity measured in the presence of 0.1 mM physiological concentration of glucose 6-phosphate and the total glycogen synthase D activity measured in the presence of 10 mM glucose 6-phosphate were significantly reduced in both pancreatic cancer groups compared with controls (Fig. 3A⇓, Table 4⇓). Fractional velocity (FV0.1), which is the percentage of glycogen synthase in the active form, was calculated as glycogen synthase I activity divided by glycogen synthase D activity. The FV0.1 was significantly lower in pancreatic cancer patients with diabetes than controls (Table 4⇓). The concentration of glucose 6-phosphate for half-maximal glycogen synthase activity was higher in pancreatic cancer patients with diabetes, however, this difference was not statistically significant (Table 4⇓). The glycogen synthase mRNA content was significantly decreased in pancreatic cancer patients with diabetes compared with controls (Fig. 3B⇓).

A, skeletal muscle glycogen synthase activity in pancreatic cancer patients and control subjects. Diluted muscle crude homogenate (30 μl) was incubated with 60 μl of UDP-[14C] glucose (∼30,000 cpm; 0.13 mM) and glycogen (6.7 g/l) in the absence and presence of various concentrations of G-6-P (0.1-10 mM) for 20 min at 30 C. Glycogen synthase activity was determined as the amount of 14C incorporated into glycogen. The results are presented as the mean ± SEM. For statistical significance, see Table 4. B, Representative gel electrophoresis of competitive RT-PCR analysis of human skeletal muscle glycogen synthase mRNA. Lanes 1-5 represent glycogen synthase RT-PCR products from 1 μg total RNA in the presence of the glycogen synthase competitive cRNA mimic (Lane 1, 143 amol; Lane 2, 46 amol; Lane 3, 14 amol; Lane 4, 4.6 amol; Lane 5, 1.4 amol). The top 301-bp DNA band is a product of the glycogen synthase competitive cRNA. B, Glycogen synthase mRNA levels were quantified by conmpetitive RT-PCR. The results are presented as the mean ± SEM. *, P < 0.05.
Skeletal muscle glycogen synthase activities in pancreatic cancer patients and normal controls
Skeletal muscle glycogen phosphorylase activity and mRNA
Glycogen phosphorylase a and b activities in nondiabetic pancreatic cancer patients were not significantly different from the controls. In contrast, the muscle glycogen phosphorylase a and bactivities were significantly higher in diabetic pancreatic cancer patients than controls (Fig. 4⇓, A and B). Although skeletal muscle glycogen phosphorylase mRNA content was higher in pancreatic cancer patients with diabetes, this difference did not reach statistical significance (Fig. 4C⇓).

Skeletal muscle glycogen phosphorylase a(A) and b (B) activities of pancreatic cancer patients and normal control subjects. Diluted muscle crude homogenate (30 μl) was incubated with 60 μl[ 14C]glucose 1-phosphate (phosphorylase a: 15 mM, specific activity 50 μCi/mmol; phosphorylase b: 100 mM, specific activity 6 μCi/mmol) and glycogen (phosphorylase a, 3.4 mg/mL; phosphorylase b, 13.4 mg/mL) for 15 min at 30 C. AMP (5 mM) was added to measure phosphorylase b activity. Glycogen phosphorylase activity was determined as the amount of 14C incorporated into glycogen. The results are presented as the mean ± SEM; *, P < 0.05. C, Quantification of glycogen phosphorylase mRNA in skeletal muscle biopsies from control subjects and pancreatic cancer patients. Representative gel electrophoresis of competitive RT-PCR analysis of human skeletal muscle glycogen phosphorylase mRNA. Lanes 1-6 represent glycogen phosphorylase RT-PCR products from 1 μg total RNA in the presence of glycogen phosphorylase competitive cRNA mimic (Lane 1, 463 amol; Lane 2, 145 amol; Lane 3, 47 amol; Lane 4, 14.5 amol; Lane 5, 4.6 amol; Lane 6, 1.4 amol). The top 293-bp DNA band is an amplification product of the endogenous glycogen phosphorylase mRNA, and the bottom 223-bp band is a product of the glycogen phosphorylase competitive cRNA. Glycogen phosphorylase mRNA levels were quantified by competitive RT-PCR and presented as the mean ± SEM.
GLUT-4 protein content and mRNA
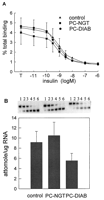
Skeletal muscle GLUT-4 protein (∼47 kDa) content was not significantly different between the cancer patients and controls (Fig. 5A⇓). Similarly, whereas GLUT-4 mRNA levels were lower in pancreatic cancer patients, no significant differences from controls were found (Fig. 5B⇓).

A, Western blot analysis of GLUT-4 content in skeletal muscles from pancreatic cancer patients and normal controls. Crude muscle membrane preparations (50 μg) were subjected to 10% SDS-PAGE. Separated proteins were transferred to nitrocellulose membrane and immunoblotted with 1F8 monoclonal antibody and 125I-labeled sheep antimouse antibody. The specific GLUT-4 band was detected and quantified by phosphorimager. B, Quantification of GLUT-4 mRNA in skeletal muscle biopsies from control subjects and pancreatic cancer patients by competitive RT-PCR. Lanes 1-6 represent GLUT-4 RT-PCR products from 1 μg total RNA in the presence of GLUT-4 competitive cRNA mimic (Lane 1, 142 amol; Lane 2, 45 amol; Lane 3, 14 amol; Lane 4, 4.5 amol; Lane 5, 1.4 amol; Lane 6, 0.5 amol). The top 315-bp DNA band is an amplification product of the endogenous GLUT-4 mRNA, and the bottom 227-bp DNA band is a product of the GLUT-4 competitive cRNA. GLUT-4 mRNA levels in skeletal muscles were quantified by competitive RT-PCR, and the results are presented as the mean ± SEM.
Discussion
There is a high incidence of diabetes and glucose intolerance in pancreatic cancer patients (1, 2). This diabetes is characterized by high basal plasma insulin concentrations, reduced whole body insulin sensitivity, and peripheral insulin resistance (2, 6, 7, 8). Skeletal muscle is the major tissue for glucose disposal and a primary site of insulin resistance (11). We have previously shown an improvement of body glucose metabolic capacity in pancreatic adenocarcinoma patients after pancreatectomy (10). In the present study, intracellular defects of insulin-regulated glucose metabolism were demonstrated in skeletal muscle from pancreatic cancer patients.
Skeletal muscle IR density, affinity, and mRNA level were normal in pancreatic cancer patients. Decreased IR binding capacity in skeletal muscle has been demonstrated in obesity (25). However, IR binding capacity and affinity are unaltered in skeletal muscle from NIDDM patients (26).
Intrinsic tyrosine kinase on the β-subunit of the IR is activated by IR interaction (27). Decreased skeletal muscle IR autophosphorylation and impaired tyrosine kinase activity have been demonstrated in obesity and NIDDM (28) and in prediabetic insulin-resistant subjects (28), suggesting that the defective receptor tyrosine kinase is an early event in the development of insulin resistance contributing to the pathophysiology of NIDDM. In contrast, IR kinase activity was normal in pancreatic cancer patients with or without diabetes, suggesting that their insulin resistance is due to postreceptor defects.
Nonoxidative glucose disposal via glycogen synthesis is a major pathway for glucose disposal in skeletal muscle, and glycogen synthase is the rate-limiting enzyme in the glycogen synthesis pathway (17). Bogardus et al. (18) have reported a correlation between skeletal muscle glycogen synthase activity and insulin-mediated glucose disposal in humans with a range of insulin sensitivities. Reduced muscle glycogen synthase activity had been reported in prediabetic and NIDDM patients (29, 30, 31). Furthermore, decreased skeletal muscle glycogen synthase protein and mRNA contents have been reported in NIDDM. (30, 32). The present study demonstrated that the insulin resistance seen in pancreatic cancer patients is associated with multiple defects of skeletal muscle glycogen synthase. Furthermore, the reduction in total glycogen synthase activity and mRNA in pancreatic cancer patients was more profound than the reductions previously reported in NIDDM (30% and 38%, respectively) (30). Insulin stimulation in skeletal muscle causes dephosphorylation and activation of glycogen synthase. The decreased fractional velocity of skeletal muscle glycogen synthase in diabetic pancreatic cancer patients suggests that the percentage of dephosphorylated active form of glycogen synthase over total enzyme is reduced, indicating an abnormal response of skeletal muscle to insulin stimulation. The glucose 6-phosphate concentration required for half-maximal glycogen synthase activation is doubled in diabetic pancreatic cancer patients, suggesting that the response of skeletal muscle glycogen synthase to its allosteric activator, glucose 6-phosphate, is also impaired. Although the nondiabetic pancreatic cancer patients had normal glucose tolerance tests, a significant reduced glucose metabolic rate has been found during hyperinsulinemic euglycemic clamp in this group, which indicates even these patients are insulin resistant (6). Although skeletal muscle glycogen synthase protein content was not determined in the present study, we observed a parallel reduction of muscle glycogen synthase mRNA with the decrease in enzyme activity. This suggests that skeletal muscle glycogen synthase gene transcription is down-regulated in pancreatic cancer patients.
Skeletal muscle glycogen content is also regulated by glycogenolysis, in which glycogen is broken down by glycogen phosphorylase. Both glycogen phosphorylase a and b activities were significantly increased in pancreatic cancer patients with diabetes, suggesting that this pathway may also play an important role in reducing muscle glycogen storage.
Insulin-regulated glucose transport via the membrane facilitative glucose transporter (GLUT-4) is considered a rate-limiting step for glucose disposal (33). Insulin-mediated glucose transport is impaired in skeletal muscle from NIDDM patients, as well as subjects with other insulin-resistant states (34). Some studies have shown no significant change of insulin-sensitive glucose transporter GLUT-4 protein content in skeletal muscles from NIDDM, suggesting that insulin-mediated glucose transport can be impaired without significant change of GLUT-4 content (35). There was no difference in distribution of GLUT-4 in either the plasma membrane of skeletal muscle or a crude membrane fraction between NIDDM patients and normal subjects (22, 36). GLUT-4 protein and mRNA contents in skeletal muscles from pancreatic cancer patients with or without diabetes were not significantly reduced from control levels. Future studies on insulin-stimulated glucose transport in skeletal muscle from pancreatic cancer patients would be of considerable interest.
In summary, multiple defects of skeletal muscle glycogen synthase activity and enhanced glycogen phosphorylase activity were seen in pancreatic cancer patients with diabetes. These observations suggest that impaired glycogen synthesis and glycogen storage is likely to be the primary site of insulin resistance in pancreatic cancer patients with diabetes. Impaired glycogen synthase activity was also found in pancreatic cancer patients without diabetes, consistent with the decreased body glucose metabolic rate in this group (6). No abnormality in skeletal muscle IR binding or receptor tyrosine kinase activity were seen, suggesting the insulin resistance associated with pancreatic cancer patients is due to defects at the post-IR level.
Improvement of insulin resistance and diabetes after tumor resection in pancreatic cancer patients suggest that these metabolic changes are in some way caused by the tumor (10). Furthermore, identical changes in muscle in the hamster ductal pancreatic adenocarcinoma model suggests that these changes in glucose metabolism are an early feature of pancreatic cancer (37). We have identified two tumor-derived factors that may influence metabolism in these patients (38, 39). One of these increases production of amylin from islet cells (38), and the other stimulates anaerobic glucose metabolism in skeletal muscle cells, causing an increase in lactate production characteristic of muscle in cancer patients (39). These and other factors may play a role in the abnormal glucose metabolism that accompanies pancreatic cancer.
Acknowledgments
We are grateful for the valuable methodological advice for the insulin receptor studies from Dr. Maria G. Buse (Department of Medicine, Medical University of South Carolina, Charleston, SC).
Footnotes
-
↵1 These studies were funded in part by the State of Nebraska Cancer and Smoking-related Diseases Program (Grant LB595) and by the Swedish Cancer Society (Grants 3450-B95-03xcc 2870-B96-06xac).
- Received May 3, 1999.
- Revision received November 2, 1999.
- Accepted November 9, 1999.
References
- ↵
Permert J, Ihse I, Jorfeldt L, von Schench H, Arnqvist HJ, Larsson J. 1993Pancreatic cancer is associated with impaired glucose metabolism. Eur J Surg. 159:101-107.
- ↵
- ↵
- ↵
Gullo L, Pezzilli R, Morselli-Labate A. 1994 Diabetes and the risk of pancreatic cancer. N Engl J Med. 79:1223-1231.
- ↵
Vecchis CL, Negri E, Franceschi S, D’Avanzo B, Boyle P. 1994 Diabetes and pancreatic cancer risk. Int J Pancretol. 16:81-84.
- ↵
- ↵
Gullo L, Anocna D, Pezzilli R, Casadei R, Campione O. 1993 Glucose tolerance and insulin secretion in pancreatic cancer. Ital J Gastroenterol. 25:487-489.
- ↵
Fogar P, Basso D, Panozzo MP, et al. 1993 C-peptide pattern in patients with pancreatic cancer. Anticancer Res.13: 2577-2580.
- ↵
Cersosimo E, Pisters P, Pesola G, McDermott K, Bajorunas D, Brennan MF. 1991Insulin secretion and action in patients with pancreatic cancer. Cancer. 67:468-493.
- ↵
Permert J, Ihse I, Jorfeldt L, von-Schenck H, Arnquist HJ, Larsson J. 1993 Improved glucose metabolism after subtotal pancreatectomy for pancreatic cancer. Br J Surg.80:1047-1050.
- ↵
DeFronzo RA. 1988 The triumvirate: β-cell, muscle, liver. A Collusion responsible for NIDDM. Diabetes. 37:667-687.
- ↵
Epstein FH. 1991 Insulin resistance: mechanisms, syndromes, and implications. N Engl J Med. 325:938-948.
- ↵
- ↵
Freychet P, Roth J, Neville Jr DM. 1971 Insulin receptors in the liver: specific binding of [125I] insulin to the plasma membrane and its relation to insulin bioactivity. Proc Natl Acad Sci USA. 68:1833-1837.
- ↵
Klein HH, Matthaei S, Drenkhan M, Ries W, Scriba PC. 1991 The relationship between insulin binding, insulin activation of insulin-receptor tyrosine kinase, and insulin stimulated glucose uptake in isolated rat adipocytes. Biochem J. 27:787-792.
- ↵
Barnard BJ, Youngren JF. 1992 Regulation of glucose transport in skeletal muscle.FASEB J. 6:3238-3244.
- ↵
Lillioja S, Mott DM, Zawadzki JK, Young AA, Abbott WG, Bogardus C. 1986 Glucose storage is a major determinant of in vivo “insulin resistance” in subjects with normal glucose tolerance. J Clin Endocrinol Metab. 62:922-927.
- ↵
Bogardus CS, Lillioja S, Stone K, Mott D. 1984 Correlation between muscle glycogen synthase activity and in vivo insulin action in man. J Clin Invest. 73:1185-1190.
- ↵
Word Health Organization Expert Committee: Diabetes Mellitus. 1985 Technical report series, no 741, Geneva: WHO.
- ↵
Bak JF, Jacobsen UK, Jorgensen FS, Pedersen O. 1989 Insulin receptor function and glycogen synthase activity in skeletal muscle biopsies from patients with insulin-dependent diabetes mellitus: effects of physical training. J Clin Endocrinol Metab. 69:158-164.
- ↵
- ↵
Lund S, Vestergaard H, Andersen PH, Schmitz O, Gotzsche LBH, Pedersen O. 1993Glut-4 content in plasma membrane of muscle from patients with non-insulin-dependent diabetes mellitus. Am J Physiol. 265:E889-E897.
- ↵
Koranyi L, Tanizawa Y, Penicaud L, Atef N, Girard J, Permutt MA. 1992Developmental regulation of amylin and insulin-gene expression in lean (Fa/Fa) and obese (fa/fa) Zuker rats. Diabetes. 41:685-690.
- ↵
Sambrook J, Fritsch EF, Maniatis T. 1989 Plasmid DNA. In: Molecular cloning, a laboratory manual, 2nd ed. Plainview, NY: Cold Spring Harbor Laboratory Press; 1.25-1.32.
- ↵
Wigand JP, Blackard WG. 1979 Down-regulation of insulin receptors in obese man.Diabetes. 28:287-291.
- ↵
Caro JF, Ittoop O, Poreis WJ. 1987 Insulin receptor kinase in human skeletal muscle from obese subjects with and without non-insulin-dependent diabetes mellitus. J Clin Invest.82:1398-1406.
- ↵
Kasuga M, Fujita-Yamaguchi Y, Blithe DI, Kahn CR. 1985 Tyrosine-specific protein kinase activity is associated with the purified insulin receptor. Proc Natl Acad Sci USA.80:2137-2141.
- ↵
- ↵
Thorburn AW, Gumbiner B, Bulacan F, Brechtel G, Henry RR. 1991 Multiple defects in muscle glycogen synthase activity contribute to reduced glycogen synthesis in non-insulin dependent diabetes mellitus. J Clin Invest. 87:489-495.
- ↵
Vestergaard H, Lund S, Larsen FS, Bjerrum O, Pedersen O. 1993 Glycogen synthase and phosphofructokinase protein and mRNA levels in skeletal muscle from insulin resistant patients with non-insulin-dependent diabetes mellitus. J Clin Invest. 91:2342-2350.
- ↵
Henry RR, Ciaraldi TP, Abrams-Carter L, Mudaliar S, Park KS, Nikoulina SE. 1996Glycogen synthase activity is reduced in cultured skeletal muscle cells of non-insulin-dependent diabetes mellitus subjects. J Clin Invest. 98:1231-1236.
- ↵
Vaag A, Henriksen JE, Beck-Nielsen H. 1992 Decreased insulin activation of glycogen synthase in skeletal muscles in young non-obese Caucasian first-degree relatives of patients with non-insulin-dependent diabetes mellitus. J Clin Invest. 89:782-788.
- ↵
Furler SM, Jenkins AB, Storlien LH, Kraegen EW. 1991 In vivo location of the rate-limiting step of hexose uptake in muscle and brain tissue of rats. Am J Physiol. 261:E337-E347.
- ↵
Dohm GL, Tapscott EB, Oaries WJ, et al. 1988 An in vitro human muscle preparation suitable for metabolic studies. Decreased insulin stimulation of glucose transport from morbidly obese and diabetic subjects. J Clin Invest. 82:486-494.
- ↵
Pederson O, Bak JF, Anderson PH, et al. 1990 Evidence against altered expression of GLUT1 or GLUT4 in skeletal muscle of patients with obesity or NIDDM. Diabetes. 39:865-870.
- ↵
Kennedy JW, Hirshman MF, Gervino EV, et al. Acute exercise induces GLUT4 translocation in skeletal muscle of normal human subjects and subjects with type 2 diabetes. Diabetes. 48:1192-1197.
- ↵
Liu J, Kazakoff K, Pour PM, Adrian TE. 1998 The intracellular mechanism of insulin resistance in hamster pancreatic ductal adenocarcinoma model. Pancreas. 17:359-366.
- ↵
- ↵
Articles citing this article
-
Diabetes mellitus and pancreatic cancer in a population-based case-control study in the san francisco bay area, california.Cancer Epidemiol. Biomarkers Prev. 2006 15: 1458-1463
-
Altered glucose metabolism and proteolysis in pancreatic cancer cell conditioned myoblasts: searching for a gene expression pattern with a microarray analysis of 5000 skeletal muscle genesGut 2004 53: 1159-1166
-
A White Paper: The Product of a Pancreas Cancer Think TankCancer Res. 2001 61: 4923-4932
Pingback: WeeksMD » Pancreatic Cancer the White Paper (but… they neglect to mention Corrective Cancer Care with IPT)