Dr. Weeks’ Comment:
Vitamin C and cancer – the research getting embarrasingly more convincing – effective, and cost-effective and… SAFE! Hmmm ask your oncologist to review this article!
Pharmacologic ascorbic acid concentrations selectively kill cancer cells: Action as a pro-drug to deliver hydrogen peroxide to tissues
- Qi Chen*,” ,
- Michael Graham Espey”¡,
- Murali C. Krishna”¡,
- James B. Mitchell”¡,
- Christopher P. Corpe*,
- Garry R. Buettner§,
- Emily Shacter” , and
- Mark Levine*,¶
+Author Affiliations
-
Communicated by J. E. Rall, National Institutes of Health, Bethesda, MD, August 2, 2005 (received for review June 1, 2005)
Abstract
Human pharmacokinetics data indicate that i.v. ascorbic acid (ascorbate) in pharmacologic concentrations could have an unanticipated role in cancer treatment. Our goals here were to test whether ascorbate killed cancer cells selectively, and if so, to determine mechanisms, using clinically relevant conditions. Cell death in 10 cancer and 4 normal cell types was measured by using 1-h exposures. Normal cells were unaffected by 20 mM ascorbate, whereas 5 cancer lines had EC50 values of <4 mM, a concentration easily achievable i.v. Human lymphoma cells were studied in detail because of their sensitivity to ascorbate (EC50 of 0.5 mM) and suitability for addressing mechanisms. Extracellular but not intracellular ascorbate mediated cell death, which occurred by apoptosis and pyknosis/necrosis. Cell death was independent of metal chelators and absolutely dependent on H2O2 formation. Cell death from H2O2 added to cells was identical to that found when H2O2 was generated by ascorbate treatment. H2O2 generation was dependent on ascorbate concentration, incubation time, and the presence of 0.5-10% serum, and displayed a linear relationship with ascorbate radical formation. Although ascorbate addition to medium generated H2O2, ascorbate addition to blood generated no detectable H2O2 and only trace detectable ascorbate radical. Taken together, these data indicate that ascorbate at concentrations achieved only by i.v. administration may be a pro-drug for formation of H2O2, and that blood can be a delivery system of the pro-drug to tissues. These findings give plausibility to i.v. ascorbic acid in cancer treatment, and have unexpected implications for treatment of infections where H2O2 may be beneficial.
Ascorbic acid (vitamin C, ascorbate) has a controversial history in cancer treatment (1). Observational reports described ascorbate, given in pharmacologic doses of 10 g daily, as effective in treating some cancers and in improving patient well-being (2–4). Subsequently, the same dose had no effect on patient well-being and survival in two double-blind placebo-controlled trials, and ascorbate was discarded as a treatment modality (5, 6). Recent clinical evidence, however, indicates that the role of ascorbate in cancer treatment should be examined anew (7). The originally reported observational studies used i.v. and oral ascorbate, but the subsequent double-blind placebo-controlled studies used only oral ascorbate. It was not recognized that the route of ascorbate administration might produce large differences in plasma concentrations. Recent pharmacokinetics studies in men and women show that 10 g of ascorbate given i.v. is expected to produce plasma concentrations of nearly 6 mM, which are >25-fold higher than those concentrations from the same oral dose (7–9). As much as a 70-fold difference in plasma concentrations is expected between oral and i.v. administration, depending on dose. Despite inconsistencies, some in vitro studies showed that ascorbate killed cancer cells, although mechanisms and physiologic relevance were unclear (10–12). Complementary and alternative medicine practitioners worldwide currently use ascorbate i.v. in some patients, in part because there is no apparent harm (13–15).
Given its potential safety and benefit, there is merit in investigating i.v. ascorbate as a possible novel cancer treatment modality. It is essential first to learn whether ascorbate acts as an anticancer agent in vitro, and if so, by what mechanisms. Our goals were to address the following: Does ascorbate in pharmacologic concentrations kill cancer cells, but not normal cells, using conditions that mimic i.v. use and a clinically relevant time course? Is action dependent on extracellular ascorbate, intracellular ascorbate, or both? If effective, what are the mechanisms? Can ascorbate be delivered to tissues without harm? Are there implications for other diseases?
We studied ascorbate at physiologic (0.1 mM) and pharmacologic (0.3-20 mM) concentrations using 1-h incubations to mimic clinical i.v. use (7–9). The data showed that pharmacologic concentrations of ascorbate killed cancer but not normal cells, that cell death was dependent only on extracellular but not intracellular ascorbate, and that killing was dependent on extracellular hydrogen peroxide (H2O2) formation with ascorbate radical as an intermediate. Ascorbate generated detectable levels of H2O2 in extracellular medium in the presence of trace serum protein but not in whole blood. The findings indicate that ascorbate at pharmacologic concentrations in blood may be a pro-drug for H2O2 delivery to tissues, with major therapeutic implications.
Materials and Methods
Cells and Reagents. Human Burkitt’s lymphoma cells (JLP-119) were obtained and studied as described in ref. 16. Other cell lines were purchased from American Type Culture Collection and were grown at 37°C in 5% CO2/95% air in recommended media containing 10% FBS (GIBCO). Human lymphocytes and monocytes were isolated by apheresis (17) from at least six healthy subjects and used immediately. Ascorbic acid was always buffered to pH 7.0 with sodium hydroxide and prepared immediately before use. Dehydroascorbic acid was freshly prepared (18). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was purchased from Molecular Probes and bacto-agar was from Difco. Other reagents, enzymes, and media were from general commercial sources.
Cell Death. Nuclear staining with Hoechst 33342 (Hoechst Pharmaceuticals) and propidium iodide (PI) was used for morphological assessment of apoptosis, necrosis, and pyknosis/necrosis by fluorescence microscopy as described in ref. 19. Briefly, 2.5 × 105 cells per ml were incubated with ascorbate or H2O2 for 1 h, washed with PBS, and suspended in fresh media. After 18-22 h, at least 200 cells were stained with Hoechst/PI and visualized under fluorescence microscopy.
MTT was used as a screening assay and performed as described in ref. 20. Cells in 96-well plates were treated with ascorbate (0.1-20 mM) for 1 h, washed, and incubated for an additional 24 h. The EC50 value was the concentration that reduced survival by 50%.
For colony formation on soft agar plates, cells were treated with 5 mM ascorbate for 1 h, washed, and plated. A two-layer agar system was used, and colonies were visualized after 10-14 days (21).
To determine the effects of red blood cells on ascorbate-induced cell death, red blood cells were prepared by centrifugation of heparinized human blood at 500 × g for 30 min. Human Burkitt’s lymphoma cells at 2.5 × 105 cells per ml were mixed with red blood cells, 25% or 50% hematocrit. Cell mixtures were treated with 2 mM ascorbate for 1 h. Lymphoma cells were recovered by using Vacutainer CPT tubes (Becton Dickinson) according to the manufacturer’s instructions. After washing, lymphoma cells were returned to fresh medium and assessed after 18 h by nuclear staining as above.
Quantitative Procedures. Catalase activity was determined by using Amplex Red (Molecular Probes) (22). Glutathione was detected by using 5,5”²-dithio bis-2-nitrobenzoic acid, and glutathione peroxidase activity was measured by a coupled reaction with glutathione reductase (Cayman Chemical, Ann Arbor, MI), according to the manufacturer’s instructions.
Ascorbate radical in culture media and blood was detected by using electron paramagnetic resonance (23, 24). Spectrometer (E9 series, Varian) settings were as follows: microwave power, 20 mW; modulation amplitude, 1.0 G; time constant, 0.25 s; scan range, 4 × 10 G; and scan time, 4 min. Radical quantitation was performed by using 3-carboxyproxyl as a standard (23).
Because ascorbate interferes with most peroxidase-based detection methods, H2O2 was measured by using a Clark-type oxygen electrode (5/6 Oxygraph, Gilson Medical Electronics, Middleton, WI). Oxygen evolution was measured upon introduction of catalase: 2H2O2 → 2H2O + O2. Calibration was performed with freshly prepared solutions of H2O2 (10-200 μM) (25).
Ascorbate was measured by HPLC with coulometric electrochemical detection (26). Protein was determined by using bicinchoninic acid (27). Cell volumes were determined by using a Coulter Multisizer II cell counter. Intracellular ascorbate concentrations were calculated by converting cell protein to a measured intracellular volume (18).
Results
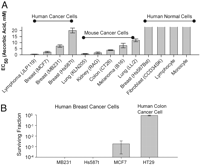
Effects of Ascorbic Acid in Pharmacologic Concentrations on Survival of Tumor and Normal Cells. We first investigated whether ascorbate in pharmacologic concentrations selectively affected the survival of cancer cells by studying nine cancer cell lines, four normal cell types, and clinically relevant conditions. Clinical pharmacokinetics analyses show that pharmacologic concentrations of plasma ascorbate, from 0.3 to 15 mM, are achievable only from i.v. administration (7). These concentrations are cleared within hours by renal filtration and excretion. In contrast, plasma ascorbate concentrations from maximum possible oral doses cannot exceed 0.22 mM because of limited intestinal absorption, which is bypassed with i.v. administration (7–9). To mimic potential clinical i.v. use, tested cells were incubated for 1 h with either pharmacologic ascorbate concentrations (0.3-20 mM) or a high physiologic concentration (0.1 mM) as control. Once ascorbate was removed, cell survival was determined by nuclear staining or MTT after 24 h (Fig. 1A). For five of the nine cancer cell lines, ascorbate concentrations causing a 50% decrease in cell survival (EC50 values) were less than 5 mM, a concentration easily achievable from i.v. infusion (7). All tested normal cells were insensitive to 20 mM ascorbate.

Fig. 1.
Effects of pharmacologic ascorbic acid concentrations on cancer and normal cells. Concentrations in this and all figures indicate final concentrations. (A) EC50 values of ascorbate in human and mouse cancer cells and normal human cells. All cells were treated with ascorbate for 1 h, washed, and recultured without ascorbate. EC50 values were determined 18-22 h later by using Hoechst/PI for human Burkitt’s lymphoma cells (JLP119), MTT and Hoechst/PI for normal lymphocytes and monocytes, and MTT for all other cells (see Materials and Methods). (B) Colony formation of cancer cells in soft agar after a 1-h treatment with 5 mM ascorbate. Surviving fraction, expressed in log scale, indicates the number of treated colonies compared with matched untreated control cells.
Colony formation assays were used as an additional means to determine cell survival (21). Four cancer cell lines were incubated with 5 mM ascorbate or untreated media for 1 h. Cells were diluted and plated and growth assessed after 14 days (Fig. 1B). All four untreated cell lines grew in soft agar, whereas three of four exposed to ascorbate displayed at least 99% growth inhibition.
Effects of Ascorbic Acid on Death of Human Lymphoma Cells. Human lymphoma cells (JLP-119) were studied in detail to determine the effects of ascorbate on cell death. Lymphoma cells were selected because of their sensitivity to ascorbate (Fig. 1A), the suitability of these cells for nuclear staining to characterize the mode of cell death (16, 19, 28), and the report of a positive clinical response of lymphoma to i.v. ascorbate (14) (unpublished work). Cells were incubated for 1 h with 0.1-5 mM ascorbate and washed, and Hoechst/PI nuclear staining was performed 18 h later to determine the amount and type of cell death (Fig. 2A). Ascorbate induced concentration-dependent cell death, which was nearly 100% at 2 mM. As ascorbate concentration increased, the pattern of death changed from apoptosis to pyknosis/necrosis, a pattern suggestive of H2O2-mediated cell death (19). We determined the time necessary for cell death after exposure to 2 mM ascorbate for 1 h (Fig. 2B). Apoptosis occurred by 6 h after exposure, and cell death by pyknosis was ≈90% at 14 h after exposure. In contrast to lymphoma cells, there was little or no killing of normal lymphocytes and monocytes by ascorbate (Fig. 2C).

Fig. 2.
Effects of ascorbic acid on human Burkitt’s lymphoma cells. Cells were treated for 1 h, washed, and recultured without ascorbate. Amounts and types of cell death were determined 18-22 h later by nuclear staining with Hoechst/PI. Types of cell death: necrosis (black), pyknosis/necrosis (gray), early apoptosis (blue), and late apoptosis (red). (A) Amount and type of cell death as a function of external ascorbate concentration. (B) Time course and type of cell death after 1 h external ascorbate (2 mM). (C) Cell death as a function of external ascorbate concentration in human Burkitt’s lymphoma cells (♦), normal lymphocytes (â–ª), and normal monocytes (â–´). (D) Cell death as a function of external ascorbate (♦) or dehydroascorbic acid (â–¡) concentrations (1-h incubation). (E) Type and amount of cell death with 2 mM ascorbate treatment, in cells previously loaded to contain 3 mM ascorbate (right), compared with unloaded cells (left).
The roles of intracellular versus extracellular ascorbate in causing cell death were examined, using ascorbate and its oxidized product dehydroascorbic acid. Ascorbate is transported into cells as such by sodium-dependent transporters, whereas dehydroascorbic acid is transported into cells by glucose transporters and then immediately reduced internally to ascorbate (29). By using either external ascorbate or external dehydroascorbic acid, lymphoma cells were loaded to equal internal concentrations of ascorbate over 1 h (data not shown). Despite similar intracellular ascorbate concentrations under both conditions, cells died only when ascorbate was present externally (Fig. 2D).
Similar to most cultured cells, lymphoma cells contain no ascorbate unless the vitamin is added to the extracellular medium (data not shown) (17). In contrast, excepting red blood cells, all cells in vivo or acutely isolated contain ascorbate, usually in millimolar concentrations. We investigated whether the prior presence of intracellular ascorbate affected death mediated by extracellular ascorbate. Lymphoma cells were preloaded with physiologic concentrations of ascorbate to produce millimolar intracellular concentrations, similar to normal lymphocytes (8, 9). Their response to external ascorbate was compared with unloaded cells (Fig. 2E). Whether or not intracellular ascorbate was preloaded, extracellular ascorbate induced the same amount and type of death. Taken together, the data in Fig. 2 A–E indicate that extracellular ascorbate in pharmacologic concentrations mediates death of lymphoma cells by apoptosis and pyknosis/necrosis, independently of intracellular ascorbate.
Mechanism of Ascorbate-Mediated Cell Killing. To determine the mechanism of ascorbate-mediated lymphoma cell death, we tested the effects of the membrane-impermeant H2O2-scavenger catalase, the membrane-permeant H2O2-scavenger tetrakis (4-benzoic acid) meso-substituted manganoporphyrin (MnTBAP) (30), and the thiol-reducing agent Tris (2-carboxyethyl) phosphine hydrochloride (TCEP) (31). We also tested whether adventitious transition metals were responsible, by using the membrane impermeant chelator diethylenetriamine-pentaacetic acid (DTPA) (32) and the membrane permeant chelator N,N”²-bis(2-hydroxybenzyl)ethylenediamine-N,N”²-diacetic acid (HBED) (33–35) (Fig. 3A). The H2O2 scavengers were completely protective, identifying H2O2 as the effector species mediating pharmacologic ascorbate-induced cell death. The effect of ascorbate was not due to chelatable, trace redox-active metals, because the two chelators had no effect on preventing death. Superoxide dismutase was not protective (data not shown), consistent with its action in producing but not degrading H2O2 (36).

Fig. 3.
Extracellular ascorbate kills human Burkitt’s lymphoma cells by generating H2O2. Cell death determined and symbolized as in Fig. 2; H2O2 measured by oxygen electrode (see Materials and Methods). (A) Effects of reactive oxygen species quenchers/scavengers, reducing agent, and metal chelators on ascorbate-mediated cell death. The following (final concentrations) were preincubated with cells for 30 min before exposure to ascorbate (2 mM): catalase (100 μg/ml); tetrakis (4-benzoic acid) meso-substituted manganoporphyrin (MnTBAP) (100 μM); Tris (2-carboxyethyl) phosphine hydrochloride (TCEP) (500 μM); diethylene-triamine-pentaacetic acid (DTPA) (1 mM); and N,N”²-bis(2-hydroxybenzyl)ethyl-enediamine-N,N”²-diacetic acid (HBED) (50 μM). (B) Type and amount of cell death as function of added H2O2 (final concentrations). (C) Cell death as a function of added H2O2 for 1 h (♦) or mean H2O2 concentration generated by 0.2-2 mM ascorbate during a 1-h incubation (â–µ). (D) Cell death in human Burkitt’s lymphoma cells (♦), normal lymphocytes (â–ª), and normal monocytes (â–´) as function of added H2O2 (final concentrations).
Because these data implicated H2O2 in cell killing, we added H2O2 to lymphoma cells and studied death patterns using nuclear staining (19, 28). The death patterns found with exogenous H2O2 exposure were similar to those found with ascorbate. For both ascorbate and H2O2, death changed from apoptosis to pyknosis/necrosis as concentrations increased (Fig. 3B).
As a specific test of ascorbate action, the amount of H2O2 formed in the presence of ascorbate was measured by using an oxygen electrode. We compared the effects on cell death of H2O2 amounts formed in the presence of ascorbate to effects from exogenously added H2O2. H2O2 generated by ascorbate oxidation and exogenously added H2O2 produced cell death curves that were indistinguishable (Fig. 3C).
Sensitivity to direct exposure to H2O2 was greater in lymphoma cells compared with normal lymphocytes and normal monocytes (Fig. 3D), consistent with the cytotoxicity pattern found above with pharmacologic ascorbate exposure. Taken together, these data are consistent with the conclusion that extracellular ascorbate induced cell death by formation of H2O2.
We investigated whether activities of intracellular H2O2-removal systems correlated with ascorbate-mediated cell death, for all cells studied. There was no association between the EC50 for ascorbate-mediated cell death and intracellular glutathione concentrations, catalase activity, or glutathione peroxidase activity (data not shown).
Mediators and Inhibitors of H2O2 Generation. H2O2 concentrations generated by ascorbate were similar with tumor cells, normal cells, or in medium without cells (data not shown), as measured by using an oxygen electrode as above. H2O2 generation was dependent on time, ascorbate concentration, and the presence of trace amounts of serum in media (Fig. 4 A and B).
Enhancing factors for ascorbate-mediated H2O2 generation in cell culture medium. H2O2 was measured by oxygen electrode, and ascorbate radical was measured by electron paramagnetic resonance. (A) H2O2 formation as function of time and ascorbate concentration: 0.2 mM (×), 0.5 mM (▴), 1 mM (□), and 2 mM (♦). (B) H2O2 formation as a function of the percentage of FBS for 1 h (2 mM ascorbate). (C) H2O2 formation as a function of ascorbate radical formation (0.2-2 mM ascorbate, 1-h incubation).
Based on these data, the most cogent explanation of ascorbate action in forming H2O2 is that the first step is ascorbate oxidation to its radical. We measured H2O2 concentration as a function of ascorbate radical concentration and found a linear relationship (Fig. 4C). These data imply that ascorbate radical is a surrogate marker for H2O2 formation.
For ascorbate to be useful clinically, it should increase the steady-state concentration of H2O2 in the extracellular milieu but not in blood. We predicted that steady-state concentrations of H2O2 generated by ascorbate oxidation would be undetectable in blood for several reasons. First, if any ascorbate radical is generated in blood, only very low concentrations are expected, and such concentrations should be lower than that needed to form detectable steady-state concentrations of H2O2 (37). Second, whatever H2O2 is generated should be removed by glutathione peroxidase and catalase within red blood cells, because H2O2 is membrane permeable (38–41). These predictions were explored in the following experiments. First, ascorbate (0-10 mM) was added to whole blood and to medium, and ascorbate radical was measured by electron paramagnetic resonance. Ascorbate radical in whole blood was not detectable when ascorbate concentrations were <3 mM and was present at minimal concentrations thereafter. In contrast, there was robust ascorbate radical generation in medium, a surrogate for extracellular fluid (Fig. 5A). Second, as direct tests, H2O2 concentrations were measured under the following conditions: In whole blood in the presence of varying concentrations of ascorbate, in whole blood after exogenous H2O2 addition, and in medium with varying concentrations of ascorbate (Fig. 5B). H2O2 was not detected in whole blood under either condition, even in the presence of far higher added concentrations than could be generated by ascorbate oxidation. Control formation of H2O2 as a function of ascorbate concentration in medium proceeded as expected. These data indicate that even if ascorbate radical was formed in blood and H2O2 was generated, it would be immediately scavenged to concentrations below detection limits. Based on these data, an additional functional experiment was conducted, based on the prediction that blood would protect tumor cells from ascorbate-mediated cell death. Lymphoma cells were incubated in the presence or absence of red blood cells, with and without added ascorbate. Red blood cells completely protected lymphoma cells from ascorbate-mediated cell death (Fig. 5C). Taken together, these data indicate that ascorbate cannot generate sustainable H2O2 concentrations in whole blood. The data are consistent with the hypothesis that ascorbate in pharmacologic concentrations is a pro-drug for H2O2 generation in the extracellular milieu but not in blood.

View larger version:
Human blood inhibits H2O2 and ascorbate radical generation from ascorbate. Ascorbate radical was measured by electron paramagnetic resonance, H2O2 was measured by oxygen electrode, and cell death was measured and displayed as in Fig. 2. (A) Ascorbate radical formation as function of ascorbate concentrations added to blood (♦) or medium (â–´). (B) H2O2 generated by ascorbate concentrations added to blood (♦) or medium (â–´) (1-h incubation), and H2O2 measured in blood immediately after the addition of indicated concentrations (â–µ). (C) Human Burkitt’s lymphoma cell death in the presence or absence of red blood cells (RBC) at 25% or 50% hematocrit (HCT) (2 mM ascorbate, 1-h treatment).
Discussion
Our data show that ascorbic acid selectively killed cancer but not normal cells, using concentrations that could only be achieved by i.v. administration and conditions that reflect potential clinical use. The effect was due only to extracellular and not intracellular ascorbate, consistent with clinical i.v. dosing. Ascorbate-mediated cell death was due to protein-dependent extracellular H2O2 generation, via ascorbate radical formation from ascorbate as the electron donor. Like glucose, when ascorbate is infused i.v., the resulting pharmacologic concentrations should distribute rapidly in the extracellular water space (42). We showed that such pharmacologic ascorbate concentrations in media, as a surrogate for extracellular fluid, generated ascorbate radical and H2O2. In contrast, the same pharmacologic ascorbate concentrations in whole blood generated little detectable ascorbate radical and no detectable H2O2. These findings can be accounted for by efficient and redundant H2O2 catabolic pathways in whole blood (e.g., catalase and glutathione peroxidase) relative to those in media or extracellular fluid (38–41). The totality of the data are consistent with the interpretation that ascorbic acid administered i.v. in pharmacologic concentrations may serve as a pro-drug for H2O2 delivery to the extracellular milieu, but without H2O2 accumulation in blood.
Although it is possible that H2O2 might accumulate in blood, this would occur only under specific conditions that reflect on the general safety of i.v. ascorbate. Ascorbate administered i.v. is likely to be safe in most patients, with virtually no toxicity compared to most currently available cancer chemotherapeutic agents. The occurrence of one predicted complication, oxalate kidney stones, is controversial (13). In patients with glucose-6-phosphate dehydrogenase deficiency, i.v. ascorbate is contraindicated because it causes intravascular hemolysis (13). The mechanism of this previously unexplained observation is now straightforward, based on the results here. H2O2 generated in blood is normally removed by catalase and glutathione peroxidase within red blood cells, with internal glutathione providing reducing equivalents. The electron source for glutathione is NADPH from the pentose shunt, via glucose-6-phosphate dehydrogenase. If activity of this enzyme is diminished, the predicted outcome is impaired H2O2 removal causing intravascular hemolysis, the observed clinical finding.
Ascorbate as a potential cancer therapeutic agent has a controversial and emotionally charged past (1, 3–6). Clinical observational studies reported possible benefit in selected patients, but double-blind placebo-controlled studies reported no benefit, and ascorbate was discarded as a potential therapy by conventional practitioners. Only recently has it been understood that the discordant clinical findings can be explained by previously unrecognized fundamental pharmacokinetics properties of ascorbate (7). In vitro effects of ascorbate on death and survival of cell lines have been reported, but there are multiple experimental concerns. For example, reports compared an experimental condition to that with no ascorbate at all (43, 44), but such a condition has had unclear physiologic relevance, because ascorbate outside and inside cells is always present unless there is severe scurvy. It was unclear whether observed effects were due to extracellular or intracellular ascorbate, or both (12, 43–46). Some experiments have used widely varying incubation times and ascorbate concentrations that have had no corresponding clinical context, making interpretation difficult. H2O2 generation by ascorbate oxidation in culture media was variously interpreted as artifact (47, 48), even though chelators had no effect (49), or reported to mediate damage internally due to diminished intracellular ascorbate, but using an H2O2 assay in which ascorbate could interfere (43, 44).
The experiments presented here provide a clear clinical context for ascorbate action. Conditions were selected to reflect peak ranges of i.v. ascorbate concentrations, which clinically might last a few hours at most, depending on the infusion rate (7). Intracellular transport of ascorbate is tightly controlled in relation to extracellular concentration (8, 9, 29). Intravenous ascorbate infusion is expected to drastically change extracellular but not intracellular concentrations (8, 9). For i.v. ascorbate to be clinically useful in killing cancer cells, pharmacologic but not physiologic extracellular concentrations should be effective, independent of intracellular ascorbate concentrations. This was what was observed here. The experiments here provide a cohesive explanation for ascorbate action in generating H2O2 outside cells, without H2O2 accumulation in blood, leading to the conclusion that ascorbate at pharmacologic concentrations in blood is a pro-drug for H2O2 delivery to tissues.
We observed that H2O2 generation was independent of metal chelators and dependent on at least 0.5% extracellular protein. The responsible proteins were between 10 and 30 kDa (data not shown). It is reasonable that extracellular milieu contains these proteins, given that extracellular milieu protein is as much as 20% of serum protein, and favors lower-molecular-weight proteins (50). Although identities of the proteins responsible are unknown, we postulate that they may have redox-active metal centers. While chelators may marginally affect these metals, they could participate in the oxidation of ascorbate when it is at pharmacologic concentrations, with subsequent formation of superoxide and H2O2 (34). It is also possible that in vivo, cell membranes and their associated proteins could harbor metals accessible to extracellular fluid and could react similarly. In either case, ascorbate, an electron-donor in such reactions, ironically initiates pro-oxidant chemistry and H2O2 formation (34, 51).
It is unknown why ascorbate, via H2O2, killed some cancer cells but not normal cells. There was no correlation with ascorbate-induced cell death and glutathione, catalase activity, or glutathione peroxidase activity. The data here showed that ascorbate initiated H2O2 formation extracellularly, but H2O2 targets could be either intracellular or extracellular, because H2O2 is membrane permeant (38, 52). For example, extracellular H2O2 might target membrane lipids, forming hydroperoxides or reactive intermediates that are quenched or repaired in normal cells but not in sensitive cancer cells. In sensitive but not resistant cancer cells, intracellular H2O2 could target DNA, DNA repair proteins, or mitochondria because of diminished superoxide dismutase activity (53). New insights may follow from future studies of a very broad range of tumor cells or from microarray analysis of resistant and sensitive cells derived from the same genetic lineage.
H2O2, as the product of pharmacologic ascorbate concentrations, has potential therapeutic uses in addition to cancer treatment, especially in infections. H2O2 is a potent mammalian antimicrobial defense mechanism (54). Neutrophils generate H2O2 from superoxide, in turn formed by NADPH oxidase-catalyzed reduction of molecular oxygen. There may be particular therapeutic application in patients with chronic granulomatous disease who have diminished superoxide production (55). Old observational animal experiments, although uncontrolled, suggest that i.v. ascorbate is effective in some viral infections (56, 57). This finding is also consistent with in vitro experiments, in which H2O2 is toxic to hepatitis C (58). Use of ascorbate as an H2O2-delivery system against sensitive pathogens, viral or bacterial, has substantial clinical implications that deserve rapid exploration.
To proceed clinically in potential treatment of infectious diseases and cancer, clear safety documentation of i.v. ascorbate administration is necessary. More than 100 patients have been described, presumably without glucose-6-phosphate dehydrogenase deficiency, who received 10 g or more of i.v. ascorbate with no reported adverse effects other than tumor lysis (3, 4, 15, 59). However, these descriptions lack formal safety documentation. Complementary and alternative medicine practitioners worldwide currently use ascorbate i.v. in doses as high as 70 g over several hours (14, 15, 59). Because i.v. ascorbate is easily available to people who seek it, a phase I safety trial in patients with advanced cancer is justified and underway.
Acknowledgments
This work was supported in part by the Intramural Research Programs of the National Institute of Diabetes and Digestive and Kidney Diseases and the National Cancer Institute (National Institutes of Health Grant Z01 DK 54506).
Footnotes
-
↵ ¶ To whom correspondence should be addressed at: Molecular and Clinical Nutrition Section, National Institutes of Health, Building 10, Room 4D52, MSC-1372, Bethesda, MD 20892-1372. E-mail: markl@mail.nih.gov.
-
Abbreviations: MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; PI, propidium iodide.
- Copyright © 2005, The National Academy of Sciences
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
-
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
-
-
- ↵
- ↵
- ↵
- ↵
-
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
Articles citing this article
-
- N. PHILIPS,
- L. DULAJ,
- and T. UPADHYA
Cancer Cell Growth and Extracellular Matrix Remodeling Mechanism of Ascorbate; Beneficial Modulation by P. leucotomosAnticancer Res 2009 29:3233–3238
-
- S. OHNO,
- Y. OHNO,
- N. SUZUKI,
- G.-I. SOMA,
- and M. INOUE
High-dose Vitamin C (Ascorbic Acid) Therapy in the Treatment of Patients with Advanced CancerAnticancer Res 2009 29:809–815
-
- L. J. Hoffer,
- M. Levine,
- S. Assouline,
- D. Melnychuk,
- S. J. Padayatty,
- K. Rosadiuk,
- C. Rousseau,
- L. Robitaille,
- and W. H. Miller
Phase I clinical trial of i.v. ascorbic acid in advanced malignancyAnn Oncol 2008 19:1969–1974
-
- Q. Chen,
- M. G. Espey,
- A. Y. Sun,
- C. Pooput,
- K. L. Kirk,
- M. C. Krishna,
- D. B. Khosh,
- J. Drisko,
- and M. Levine
From the Cover: Pharmacologic doses of ascorbate act as a prooxidant and decrease growth of aggressive tumor xenografts in miceProc. Natl. Acad. Sci. USA 2008 105:11105–11109
-
- B. Frei
- and S. Lawson
Vitamin C and cancer revisitedProc. Natl. Acad. Sci. USA 2008 105:11037–11038
-
- L. J. Hoffer,
- M. Levine,
- S. Assouline,
- D. Melnychuk,
- S. J. Padayatty,
- K. Rosadiuk,
- C. Rousseau,
- L. Robitaille,
- and W. H. Miller
Phase I clinical trial of i.v. ascorbic acid in advanced malignancyAnn Oncol 2008 0:mdn377v4–mdn377
-
- Y. Li
- and H. E. Schellhorn
New Developments and Novel Therapeutic Perspectives for Vitamin CJ. Nutr. 2007 137:2171–2184
-
- S. Ohtani,
- A. Iwamaru,
- W. Deng,
- K. Ueda,
- G. Wu,
- G. Jayachandran,
- S. Kondo,
- E. N. Atkinson,
- J. D. Minna,
- J. A. Roth,
- and L. Ji
Tumor Suppressor 101F6 and Ascorbate Synergistically and Selectively Inhibit Non-Small Cell Lung Cancer Growth by Caspase-Independent Apoptosis and AutophagyCancer Res. 2007 67:6293–6303
-
- Q. Chen,
- M. G. Espey,
- A. Y. Sun,
- J.-H. Lee,
- M. C. Krishna,
- E. Shacter,
- P. L. Choyke,
- C. Pooput,
- K. L. Kirk,
- G. R. Buettner,
- and M. Levine
Ascorbate in pharmacologic concentrations selectively generates ascorbate radical and hydrogen peroxide in extracellular fluid in vivoProc. Natl. Acad. Sci. USA 2007 104:8749–8754
-
- B. Halliwell
Dietary polyphenols: Good, bad, or indifferent for your health?Cardiovasc Res 2007 73:341–347
-
- E. J. Meredith,
- M. J. Holder,
- A. Rosen,
- A. D. Lee,
- M. J. S. Dyer,
- N. M. Barnes,
- and J. Gordon
Dopamine targets cycling B cells independent of receptors/transporter for oxidative attack: Implications for non-Hodgkin”s lymphomaProc. Natl. Acad. Sci. USA 2006 103:13485–13490
-
- S. Assouline
- and W. H. Miller
High-dose vitamin C therapy: renewed hope or false promise?CMAJ 2006 174:956–957
-
- K. I. Block
Why Integrative Therapies?Integr Cancer Ther 2006 5:3–6
-
- R. W. Moss
Should Patients Undergoing Chemotherapy and Radiotherapy Be Prescribed Antioxidants?Integr Cancer Ther 2006 5:63–82
-
- M. J. Gonzalez
- and J. R. Miranda-Massari
Advances in Vitamin C ResearchIntegr Cancer Ther 2006 5:7–8
-
- K. I. Block
Antioxidants in the NewsIntegr Cancer Ther 2005 4:271–273
-
- K. I. Block
- and C. Gyllenhaal
Commentary: The Pharmacological Antioxidant Amifostine–Implications of Recent Research for Integrative Cancer CareIntegr Cancer Ther 2005 4:329–351