Dr. Weeks’ Comment: Statin drugs are given out like candy and in all instances, they compromise the benefits of naturaly occurring energy agents like CoQ 10 and in most instances the statin over-prescription results in deleterious and worrisome side-effects. (In fact the patent for statins INCLUDED Co Q 10 but Big Pharma ignored that caveat!) Prior posts clarify that cholesterol is much less dangerous than you are led to believe (it is the oxidized cholesterol which is dangerous – ask your doctor to do this test – thanks Duke!) Additionally, these posts implicate statins in problems related to memory loss (reported by the Wall Street Journal!) Alzheimer’s disease, increased risk of heart failure, Parkinson’s disease and many more illnesses! Now we see that statin drugs help primarily because they offer anti-inflammation. But anti-inflammation at what a horrible cost! Statin drugs fatigue all critical systems when they destroy Co Q 10 – like unplugging your battery charger and all systems wear down!
Most people reading this website use SOUL instead. If you choose an anti-inflammatory agent, try SOUL (money back guarantee…. eat 29 of the 30 packets and if not satisfied, RAIN company will refund the cost of product!)
It is the most “centsible” (safe, effective and cost effective) anti-inflammatory agent out there!
Are statins anti-inflammatory?
Gavin J Blake and Paul M Ridker
Abstract
Large scale clinical trials demonstrate significant reductions in cardiovascular event rates with statin therapy. The observed benefit of statin therapy, however, may be larger in these trials than that expected on the basis of lipid lowering alone. Emerging evidence from both clinical trials and basic science studies suggest that statins have anti-inflammatory properties, which may additionally lead to clinical efficacy. Measurement of markers of inflammation such as high sensitivity C-reactive protein in addition to lipid parameters may help identify those patients who will benefit most from statin therapy.
Introduction
Hyperlipidemia is a major risk factor for atherosclerosis, and several large scale trials demonstrate that cholesterol lowering therapy with 3-hydroxy-3-methyl coenzyme A (HMGCoA) reductase inhibitors reduces coronary event rates in both primary and secondary prevention [1,2,3,4,5]. Paradoxes revealed by these trials, however, raise the possibility that statins may have effects that go beyond simple lipid reduction.
Despite large reductions in cardiac event rates, the absolute angiographic change in arterial narrowing observed with statin therapy is small [6]. Second, several trials suggest that the observed clinical benefit of statin therapy is greater than that expected on the basis of low density lipoprotein (LDL) reduction alone. For instance, when the Framingham coronary heart disease model was applied to the West of Scotland Coronary Prevention Study (WOSCOPS) population, the model accurately predicted risk in the placebo group but underestimated the risk reduction in the pravastatin group [2]. The benefits of LDL reduction with statins also appear to occur earlier than is observed with other cholesterol lowering therapies such as cholestyramine and ileal bypass surgery, even among patients with similar levels of cholesterol after therapy [7,8]. Finally, statins reduce the risk of stroke, but LDL is not an important risk factor for stroke [9].
One additional mechanism by which statins may reduce vascular event rates relates to potential anti-inflammatory effects of these agents. Inflammatory processes, in this regard, play a pivotal role in the pathogenesis of atherosclerosis, and elevated plasma levels of markers of inflammation such as high sensitivity C-reactive protein (hs-CRP), serum amyloid A, IL-6 and soluble intercellular adhesion molecule-1 have been shown to predict cardiovascular events [10,11,12,13,14,15,16].
Laboratory evidence for anti-inflammatory effects of statins
Unstable plaques are characterized by active inflammation that overwhelms the plaque’s capacity for repair [17]. Macrophages and T cells are abundant in the regions of plaque rupture, while smooth muscle cells are few. Stable plaques, conversely, contain few inflammatory cells and abundant smooth muscle cells.
Numerous studies suggest important effects of statins on macrophage function. Macrophages are capable of degrading the extracellular matrix and, by secreting matrix metalloproteinase (MMP), may weaken the fibrous cap and thus predispose an atheromatous plaque to rupture. Fluvastatin and simvastatin have recently been shown to inhibit MMP-9 (gelatinase B) activity and secretion by macrophages [18]. This effect is reversed by the addition of mevalonate, suggesting that it is mediated by HMGCoA reductase inhibition.
MMP-1, or interstitial collagenase, is also thought to play a role in atherosclerotic plaque rupture. Fluvastatin appears to decrease MMP-1 expression in human vascular endothelial cells in a time- and dose-dependent manner [19]. This effect is also seen with lovastatin and again is completely blocked by coincubation with mevalonate. The concentration of fluvastatin required to reduce MMP-1 expression is similar to that seen in clinical practice.
Pravastatin has been shown to cause changes in the composition of atheromatous plaque independent of its cholesterol lowering effect. Pravastatin-treated monkeys have better vasodilator function and favorable changes in the composition of atheromatous plaque compared with control animals with similar changes in lipid profile caused by diet alone [20]. The pravastatin-treated monkeys had fewer macrophages in the intima and media, less calcification and less neovascularization in the intima. Pravastatin may thus serve to stabilize vulnerable plaques by promoting regression of fragile, rupture prone microvessels in the intima.
Oxidized LDL is a key player in the atherogenic pathway. The uptake of oxidized LDL by macrophages generates lipid rich foam cells. Oxidized LDL causes monocyte tissue factor expression, and the proliferation and apoptosis of smooth muscle cells [21,22]. Oxidized LDL also inhibits nitric oxide synthase activity and hence impairs endothelium-dependent vasodilation [23].
Statins reduce the susceptibility of LDL to oxidation by a variety of mechanisms. Statins reduce the cholesterol content of lipoproteins through their hypocholesterolemic effects, and thus lower the amount of substrate available for oxidation [24]. Simvastatin has been shown to reduce macrophage superoxide formation, thereby decreasing cell oxygen production [25]. Fluvastatin and lovastatin bind to phospholipid on the surface of LDL and thus prevent diffusion into the lipoprotein core of free radicals generated under oxidative stress [26]. Atorvastatin and fluvastatin have also been shown to have direct antioxidant potential [27,28].
Statins can directly upregulate endothelial nitric oxide synthase (eNOS) expression in vitro under cholesterol clamped conditions [23]. Both simvastatin and lovastatin upregulate eNOS expression almost fourfold, and completely prevent its downregulation by oxidized LDL. The upregulation of eNOS was reversed by the addition of mevalonate.
A significant increase in endothelium-dependent vasodilation in patients with moderate hypercholesterolemia has been observed after 4 weeks of treatment with simvastatin [29]. The neuroprotective effect of statins is absent in eNOS deficient mice, suggesting that enhanced eNOS activity by statins is a main mechanism by which HMGCoA reductase inhibitors protect against cerebral injury [30].
Hypercholesterolemic rats treated with fluvastatin have attenuated leukocyte adherence responses to platelet activation factor and leukotriene B4 [31]. Statins inhibit the expression of CD-11b on the cell surface, thus reducing the adhesiveness of macrophages to the vascular endothelium [32]. Atorvastatin reduces monocyte chemoattractant protein-1 levels in the intima and media in hypercholesterolemic rabbits [33]. This decrease in monocyte chemoattractant protein-1 is related to a reduction in nuclear factor κB activation, a transcription factor involved in the induction of monocyte chemoattractant protein-1 and other proinflammatory cytokines such as IL-1β and tumor necrosis factor-α (TNF-α).
Statins also cause decreased macrophage expression of soluble intercellular adhesion molecule-1 and lipopolysaccharide-induced secretion of IL-6 and TNF-α by monocytes and macrophages [34,35,36]. Recent data show that simvastatin therapy for 8 weeks reduces monocyte expression of TNF-α and IL-1β by 49 and 35%, respectively [37]; this is intriguing data because elevated plasma levels of both soluble intercellular adhesion molecule-1 and IL-6 have been shown to predict risk for myocardial infarction [12,13]. A recent analysis from the Cholesterol and Recurrent Events (CARE) trial showed that plasma concentrations of TNF-α are also persistently elevated among postmyocardial infarction patients at increased risk for coronary events [38]. These findings provide supportive evidence that anti-inflammatory effects of statins may make an important contribution to their clinical efficacy.
In addition to reducing synthesis of cholesterol, HMGCoA reductase inhibitors lower levels of isoprenoids, which are derived from intermediates in the cholesterol biosynthetic pathway. Isoprenoids prenylate a number of cellular proteins that play key roles in cell growth and signal transduction pathways such as G proteins, which have been shown to modulate mitogenic pathways [39].
Statins have been reported to induce apoptosis in cultured vascular smooth muscle cells, and both atorvastatin and fluvastatin increase apoptosis in injured carotid arteries in rabbits [40]. Both simvastatin and fluvastatin inhibit smooth muscle cell proliferation, while pravastatin is devoid of such an effect [41]. The hydrophilic nature of pravastatin may thus limit its diffusion through cell membranes.
Statins also have potentially favorable effects on the coagulation profile. Tissue factor is the primary initiator of the extrinsic pathway. Lipophilic statins (simvastatin and fluvastatin) have been shown to decrease tissue factor expression and activity in cultured human monocyte derived macrophages [42]. Statins also increase tissue plasminogen activator levels and cause a concomitant fall in plasminogen activating inhibitor-1 activity [43].
Other in vivo effects common to statins include a reduction of platelet aggregation ex vivo and in vitro[44]. Simvastatin and pravastatin have been shown to reduce thrombus formation and inhibit thrombin generation [45,46].
Pravastatin therapy is associated with a reduction in the number of episodes of rejection following cardiac transplantation. The inhibition of natural killer T cell activity by pravastatin may explain, in part, this beneficial effect [47]. Although transplant vasculopathy is an entity distinct from atherosclerotic disease, similar inflammatory mediators may contribute to plaque instability.
Evidence from clinical trials for anti-inflammatory effects of statins
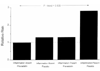
Clinical data regarding the anti-inflammatory role of statins has until recently been limited. Intriguing data from the CARE trial suggests that pravastatin may directly attenuate the adverse effects of inflammation in a process independent of LDL lowering. The CARE trial specifically randomized patients with a prior history of myocardial infarction to receive either 40 mg pravastatin daily or placebo [1]. Patients with evidence of persistent inflammation (as evidenced by elevation of both hs-CRP and serum amyloid A) were at increased risk for recurrent cardiovascular events [48]. The study group with the highest risk of recurrent events was that of patients with persistent evidence of inflammation who were assigned to placebo (relative risk=2.81, P = 0.007). In a stratified analysis, however, the association between inflammation and risk was significant among those randomized to placebo (relative risk=2.11, P = 0.048) but was attenuated and not significant among those randomized to pravastatin (relative risk=1.29, P = 0.5). The proportion of recurrent cardiac events prevented by pravastatin was 54% among those patients with persistent evidence of inflammation compared with 25% among those without inflammation (Fig. ”‹(Fig.1).1). This difference in benefit was observed despite identical baseline LDL levels in these two groups. Compared with placebo, moreover, pravastatin therapy resulted in a 22% reduction in median hs-CRP levels over a 5 year period (Fig. ”‹(Fig.2),2), an effect that was independent of statin-induced changes in LDL [49]. Taken together, these data suggest that, in addition to lowering LDL cholesterol, pravastatin may have clinically important anti-inflammatory properties.


Atorvastatin and simvastatin have also been shown to reduce CRP levels in a small study of 66 hyperlipidemic patients with coronary artery disease [50]. Simvastatin has been found to reduce CRP levels in type II diabetic patients with hyperlipidemia [51]. The observed change in CRP did not correlate with changes in total cholesterol or high density lipoprotein.
The relative importance of hs-CRP reduction compared with LDL reduction is currently uncertain, and several ongoing studies are directly addressing this issue. What is clear, however, is that markers of inflammation such as hs-CRP appear to add to the predictive value of lipid screening in terms of predicting cardiovascular risk (Fig. ”‹(Fig.3)3) [11,52]. It is also clear, at least in secondary prevention, that statins lower hs-CRP levels even in the absence of hyperlipidemia. It has thus been hypothesized that screening for inflammatory markers may provide an improved method to target statin therapy in primary prevention.

It is not currently known if all statins have clinically relevant anti-inflammatory effects or whether any one agent is more powerful than another is in this regard. Furthermore, the time course of the anti-inflammatory effects of statins is not known. Clinical trials with head to head comparison of statins (such as the PROVE-IT study) and studies designed to examine the time-course of statin therapy on hs-CRP levels (such as the PRINCE trial) will help to resolve these remaining questions [53].
Article information
 Corresponding author.
Corresponding author.References
- Sacks FM, Pfeffer MA, Moye LA, Rouleau JL, Rutherford JD, Cole TG, Brown L, Warnica JW, Arnold JM, Wun CC, Davis BR, Braunwald E. The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. Cholesterol and Recurrent Events Trial investigators. N Engl J Med. 1996;335:1001-1009. doi: 10.1056/NEJM199610033351401. [PubMed][Cross Ref]
- Shepherd J, Cobbe SM, Ford I, Isles CG, Lorimer AR, MacFarlane PW, McKillop JH, Packard CJ. Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. West of Scotland Coronary Prevention Study Group. N Engl J Med.1995;333:1301-1307. doi: 10.1056/NEJM199511163332001. [PubMed][Cross Ref]
- Downs JR, Clearfield M, Weis S, Whitney E, Shapiro DR, Beere PA, Langendorfer A, Stein EA, Kruyer W, Gotto AM., Jr Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels: results of AFCAPS/TexCAPS. Air Force/Texas Coronary Atherosclerosis Prevention Study. JAMA. 1998;279:1615-1622. doi: 10.1001/jama.279.20.1615. [PubMed] [Cross Ref]
- Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. The Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group. N Engl J Med.1998;339:1349-1357. doi: 10.1056/NEJM199811053391902. [PubMed][Cross Ref]
- Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet.1994;344:1383-1389. doi: 10.1016/S0140-6736(94)90566-5. [PubMed] [Cross Ref]
- de Groot E, Jukema JW, Montauban van Swijndregt AD, Zwinderman AH, Ackerstaff RG, van der Steen AF, Bom N, Lie KI, Bruschke AV. B-Mode ultrasound assessment of pravastatin treatment effect on carotid and femoral artery walls and its correlations with coronary arteriographic findings: a report of the Regression Growth Evaluation Statin Study (REGRESS). J Am Coll Cardiol. 1998;31:1561-1567. doi: 10.1016/S0735-1097(98)00170-3. [PubMed][Cross Ref]
- The Lipid Research Clinics Coronary Primary Prevention Trial results. I. Reduction in incidence of coronary heart disease. JAMA. 1984;251:351-364.[PubMed]
- Buchwald H, Campos CT, Boen JR, Nguyen PA, Williams SE. Disease-free intervals after partial ileal bypass in patients with coronary heart disease and hypercholesterolemia: report from the Program on the Surgical Control of the Hyperlipidemias (POSCH). J Am Coll Cardiol. 1995;26:351-357. doi: 10.1016/0735-1097(95)80006-3. [PubMed] [Cross Ref]
- Cholesterol, diastolic blood pressure, and stroke: 13,000 strokes in 450,000 people in 45 prospective cohorts. Prospective studies collaboration. Lancet.1995;346:1647-1653. doi: 10.1016/S0140-6736(95)92836-7. [PubMed] [Cross Ref]
- Ridker PM, Cushman M, Stampfer MJ, Tracy RP, Hennekens CH. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N Engl J Med. 1997;336:973-979. doi: 10.1056/NEJM199704033361401. [PubMed][Cross Ref]
- Ridker PM, Hennekens CH, Buring JE, Rifai N. C-Reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N Engl J Med. 2000;342:836-843. doi: 10.1056/NEJM200003233421202. [PubMed][Cross Ref]
- Ridker PM, Rifai N, Stampfer MJ, Hennekens CH. Plasma concentration of interleukin-6 and the risk of future myocardial infarction among apparently healthy men. Circulation. 2000;101:1767-1772.[PubMed]
- Ridker PM, Hennekens CH, Roitman-Johnson B, Stampfer MJ, Allen J. Plasma concentration of soluble intercellular adhesion molecule 1 and risks of future myocardial infarction in apparently healthy men.Lancet. 1998;351:88-92. doi: 10.1016/S0140-6736(97)09032-6. [PubMed] [Cross Ref]
- Hwang SJ, Ballantyne CM, Sharrett AR, Smith LC, Davis CE, Gotto AM, Jr, Boerwinkle E. Circulating adhesion molecules VCAM-1, ICAM-1, and E-selectin in carotid atherosclerosis and incident coronary heart disease cases: the Atherosclerosis Risk In Communities (ARIC) study. Circulation. 1997;96:4219-4225.[PubMed]
- Harris TB, Ferrucci L, Tracy RP, Corti MC, Wacholder S, Ettinger WH, Jr, Heimovitz H, Cohen HJ, Wallace R. Associations of elevated interleukin-6 and C-reactive protein levels with mortality in the elderly. Am J Med. 1999;106:506-512. doi: 10.1016/S0002-9343(99)00066-2. [PubMed] [Cross Ref]
- Danesh J, Whincup P, Walker M, Lennon L, Thomson A, Appleby P, Gallimore JR, Pepys MB. Low grade inflammation and coronary heart disease: prospective study and updated meta-analyses. BMJ.2000;321:199-204. doi: 10.1136/bmj.321.7255.199.[PMC free article] [PubMed] [Cross Ref]
- Ross R. Atherosclerosis – an inflammatory disease. N Engl J Med. 1999;340:115-126. doi: 10.1056/NEJM199901143400207. [PubMed][Cross Ref]
- Bellosta S, Via D, Canavesi M, Pfister P, Fumagalli R, Paoletti R, Bernini F. HMG-CoA reductase inhibitors reduce MMP-9 secretion by macrophages. Arterioscler Thromb Vasc Biol. 1998;18:1671-1678. [PubMed]
- Ikeda U, Shimpo M, Ohki R, Inaba H, Takahashi M, Yamamoto K, Shimada K. Fluvastatin inhibits matrix metalloproteinase-1 expression in human vascular endothelial cells. Hypertension. 2000;36:325-329.[PubMed]
- Williams JK, Sukhova GK, Herrington DM, Libby P. Pravastatin has cholesterol-lowering independent effects on the artery wall of atherosclerotic monkeys. J Am Coll Cardiol. 1998;31:684-691. doi: 10.1016/S0735-1097(97)00537-8. [PubMed][Cross Ref]
- Bjorkerud B, Bjorkerud S. Contrary effects of lightly and strongly oxidized LDL with potent promotion of growth versus apoptosis on arterial smooth muscle cells, macrophages, and fibroblasts. Arterioscler Thromb Vasc Biol. 1996;16:416-424. [PubMed]
- Broze GJ., Jr The role of tissue factor pathway inhibitor in a revised coagulation cascade. Semin Hematol. 1992;29:159-169. [PubMed]
- Laufs U, La Fata V, Plutzky J, Liao JK. Upregulation of endothelial nitric oxide synthase by HMG CoA reductase inhibitors. Circulation. 1998;97:1129-1135.[PubMed]
- Hoffman R, Brook GJ, Aviram M. Hypolipidemic drugs reduce lipoprotein susceptibility to undergo lipid peroxidation: in vitro and ex vivo studies.Atherosclerosis. 1992;93:105-113. [PubMed]
- Giroux LM, Davignon J, Naruszewicz M. Simvastatin inhibits the oxidation of low-density lipoproteins by activated human monocyte-derived macrophages.Biochim Biophys Acta. 1993;1165:335-338. doi: 10.1016/0005-2760(93)90145-Y. [PubMed][Cross Ref]
- Aviram M, Hussein O, Rosenblat M, Schlezinger S, Hayek T, Keidar S. Interactions of platelets, macrophages, and lipoproteins in hypercholesterolemia: antiatherogenic effects of HMG-CoA reductase inhibitor therapy. J Cardiovasc Pharmacol. 1998;31:39-45. doi: 10.1097/00005344-199801000-00006. [PubMed] [Cross Ref]
- Aviram M, Rosenblat M, Bisgaier CL, Newton RS. Atorvastatin and gemfibrozil metabolites, but not the parent drugs, are potent antioxidants against lipoprotein oxidation. Atherosclerosis. 1998;138:271-280. doi: 10.1016/S0021-9150(98)00032-X. [PubMed][Cross Ref]
- Suzumura K, Yasuhara M, Tanaka K, Suzuki T. Protective effect of fluvastatin sodium (XU-62-320), a 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitor, on oxidative modification of human low-density lipoprotein in vitro. Biochem Pharmacol. 1999;57:697-703. doi: 10.1016/S0006-2952(98)00341-4. [PubMed] [Cross Ref]
- O’Driscoll G, Green D, Taylor RR. Simvastatin, an HMG-coenzyme A reductase inhibitor, improves endothelial function within 1 month. Circulation.1997;95:1126-1131. [PubMed]
- Endres M, Laufs U, Huang Z, Nakamura T, Huang P, Moskowitz MA, Liao JK. Stroke protection by 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase inhibitors mediated by endothelial nitric oxide synthase. Proc Natl Acad Sci USA. 1998;95:8880-8885. doi: 10.1073/pnas.95.15.8880.[PMC free article] [PubMed] [Cross Ref]
- Kimura M, Kurose I, Russell J, Granger DN. Effects of fluvastatin on leukocyte-endothelial cell adhesion in hypercholesterolemic rats. Arterioscler Thromb Vasc Biol. 1997;17:1521-1526. [PubMed]
- Weber C, Erl W, Weber KS, Weber PC. HMG-CoA reductase inhibitors decrease CD11b expression and CD11b-dependent adhesion of monocytes to endothelium and reduce increased adhesiveness of monocytes isolated from patients with hypercholesterolemia. J Am Coll Cardiol.1997;30:1212-1217. doi: 10.1016/S0735-1097(97)00324-0. [PubMed] [Cross Ref]
- Bustos C, Hernandez-Presa MA, Ortego M, Tunon J, Ortega L, Perez F, Diaz C, Hernandez G, Egido J. HMG-CoA reductase inhibition by atorvastatin reduces neointimal inflammation in a rabbit model of atherosclerosis. J Am Coll Cardiol. 1998;32:2057-2064. doi: 10.1016/S0735-1097(98)00487-2.[PubMed] [Cross Ref]
- Niwa S, Totsuka T, Hayashi S. Inhibitory effect of fluvastatin, an HMG-CoA reductase inhibitor, on the expression of adhesion molecules on human monocyte cell line. Int J Immunopharmacol. 1996;18:669-675. doi: 10.1016/S0192-0561(96)00068-9. [PubMed][Cross Ref]
- Ikeda U, Shimada K. Statins and monocytes [letter].Lancet. 1999;353:2070. [PubMed]
- Rosenson RS, Tangney CC, Casey LC. Inhibition of proinflammatory cytokine production by pravastatin.Lancet. 1999;353:983-984. doi: 10.1016/S0140-6736(99)00564-4. [PubMed] [Cross Ref]
- Ferro D, Parrotto S, Basili S, Alessandri C, Violi F. Simvastatin inhibits the monocyte expression of proinflammatory cytokines in patients with hypercholesterolemia. J Am Coll Cardiol.2000;36:427-431. doi: 10.1016/S0735-1097(00)00771-3. [PubMed] [Cross Ref]
- Ridker PM, Rifai N, Pfeffer M, Sacks F, Lepage S, Braunwald E. Elevation of tumor necrosis factor-alpha and increased risk of recurrent coronary events after myocardial infarction. Circulation.2000;101:2149-2153. [PubMed]
- Hall A. The cellular functions of small GTP-binding proteins. Science. 1990;249:635-640. [PubMed]
- Baetta R, Donetti E, Comparato C, Calore M, Rossi A, Teruzzi C, Paoletti R, Fumagalli R, Soma MR. Proapoptotic effect of atorvastatin on stimulated rabbit smooth muscle cells. Pharmacol Res.1997;36:115-121. doi: 10.1006/phrs.1997.0211.[PubMed] [Cross Ref]
- Corsini A, Bernini F, Quarato P, Donetti E, Bellosta S, Fumagalli R, Paoletti R, Soma VM. Non-lipid-related effects of 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors. Cardiology. 1996;87:458-468.[PubMed]
- Colli S, Eligini S, Lalli M, Camera M, Paoletti R, Tremoli E. Vastatins inhibit tissue factor in cultured human macrophages. A novel mechanism of protection against atherothrombosis. Arterioscler Thromb Vasc Biol. 1997;17:265-272. [PubMed]
- Essig M, Nguyen G, Prie D, Escoubet B, Sraer JD, Friedlander G. 3-Hydroxy-3-methylglutaryl coenzyme A reductase inhibitors increase fibrinolytic activity in rat aortic endothelial cells. Role of geranylgeranylation and Rho proteins. Circ Res. 1998;83:683-690.[PubMed]
- Farnier M, Davignon J. Current and future treatment of hyperlipidemia: the role of statins. Am J Cardiol.1998;82:3J-10J. doi: 10.1016/S0002-9149(98)00423-8. [PubMed] [Cross Ref]
- Dangas G, Badimon JJ, Smith DA, Unger AH, Levine D, Shao JH, Meraj P, Fier C, Fallon JT, Ambrose JA. Pravastatin therapy in hyperlipidemia: effects on thrombus formation and the systemic hemostatic profile. J Am Coll Cardiol. 1999;33:1294-1304. doi: 10.1016/S0735-1097(99)00018-2. [PubMed][Cross Ref]
- Szczeklik A, Musial J, Undas A, Gajewski P, Gora P, Swadzba J, Jankowski M. Inhibition of thrombin generation by simvastatin and lack of additive effects of aspirin in patients with marked hypercholesterolemia. J Am Coll Cardiol.1999;33:1286-1293. doi: 10.1016/S0735-1097(99)00023-6. [PubMed] [Cross Ref]
- Kobashigawa JA, Katznelson S, Laks H, Johnson JA, Yeatman L, Wang XM, Chia D, Terasaki PI, Sabad A, Cogert GA, Trosian K, Hamilton MA, Moriguchi JD, Kawata N, Hage A, Drinkwater DC, Stevenson LW. Effect of pravastatin on outcomes after cardiac transplantation. N Engl J Med. 1995;333:621-627. doi: 10.1056/NEJM199509073331003. [PubMed][Cross Ref]
- Ridker PM, Rifai N, Pfeffer MA, Sacks FM, Moye LA, Goldman S, Flaker GC, Braunwald E. Inflammation, pravastatin, and the risk of coronary events after myocardial infarction in patients with average cholesterol levels. Cholesterol and Recurrent Events (CARE) Investigators. Circulation. 1998;98:839-844.[PubMed]
- Ridker PM, Rifai N, Pfeffer MA, Sacks F, Braunwald E. Long-term effects of pravastatin on plasma concentration of C-reactive protein. The Cholesterol and Recurrent Events (CARE) Investigators.Circulation. 1999;100:230-235. [PubMed]
- Strandberg TE, Vanhanen H, Tikkanen MJ. Effect of statins on C-reactive protein in patients with coronary artery disease. Lancet. 1999;353:118-119. [PubMed]
- Kluft C, de Maat MP, Gevers Leuven JA, Potter van Loon BJ, Mohrschladt MF. Statins and C-reactive protein [letter]. Lancet. 1999;353:1274. [PubMed]
- Ridker PM, Glynn RJ, Hennekens CH. C-Reactive protein adds to the predictive value of total and HDL cholesterol in determining risk of first myocardial infarction. Circulation. 1998;97:2007-2011. [PubMed]
- Ridker PM. Are statins anti-inflammatory? Issues in the design and conduct of the pravastatin inflammation C-reactive protein evaluation. Curr Cardiol Rep. 2000;2:269-273. [PubMed]